Efficient Alignment and Variant Calling Using Sequencing Data
Discover how fast single-pass alignment and variant calling using sequencing data can revolutionize genomic methods, reducing computational burden and improving accuracy. Learn about the motivation, sequence computation, previous strategies, benefits of the new strategy, and the algorithm used in Findmap.f90.

Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author. If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
You are allowed to download the files provided on this website for personal or commercial use, subject to the condition that they are used lawfully. All files are the property of their respective owners.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.
E N D
Presentation Transcript
Fast Single-Pass Alignment and Variant Calling Using Sequencing Data P.M. VanRaden and D.M. Bickhart Animal Genomics and Improvement Laboratory, Agricultural Research Service, USDA, Beltsville, MD, USA paul.vanraden@ars.usda.gov Plant & Animal Genome, San Diego, California January 9 -11, 2016 (1)
Motivation l Genomic methods require much computation w Genotype models replaced pedigree models w Sequence variants replacing chip genotypes w Both increased data by orders of magnitude l Fast methods are available for imputation l Slow methods for alignment, variant calling Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (2) Paul VanRaden
Sequence computation l Alignment reports the chromosome location that best matches the short (150-base) DNA segment to the reference map (2.7 billion). l Often both ends of a longer segment are read and these paired ends are located together. l Variant calling reports if each mapped segment contains a reference or alternate allele at any site. These variants could be previously known or newly discovered. Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (3) Paul VanRaden
Previous strategies l Almost all programs do alignment, and then variant calling, instead of both together. l Program BWA was examined as a popular alignment strategy, and GATK for calling. w Mapping reads to the reference is a first critical computational challenge whose cost necessitates that each read be aligned independently, guaranteeing that many reads spanning indels will be misaligned. DePristo et al (2011) GATK paper Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (4) Paul VanRaden
Benefits of new strategy l Most programs align only to Dominette s DNA l Findmap can align using all known DNA differences among and within breeds l Error rate is reduced by separating known SNPs and indels from machine read errors l Locations are mapped back to the same common reference (UMD3.1) Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (5) Paul VanRaden
Algorithm used in findmap.f90 l Read reference map, store in hash table l Read and hash known variants (SNPs & indels) l Process batches of 1 million paired end reads, send to multiple processors sharing memory w Find location where both ends match map w Count alleles (reference, alternate) & errors l Output alignment and variant call files Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (6) Paul VanRaden
Gaps, k-mers, and hashing strategy Identify long gaps between the same base (A, C, G, or T) TGGATTCTTTATCACTGAGCTACCTGGGAAGCCAAGTAAGC Extend each gap to a 16-base k-mer, convert to an 8-byte integer: Basenum (1, 2, 3, 4) = Base (A, C, G, T) Hashnum = 4 * Hashnum + Basenum , loop across 16-base k-mer Apply hash function (written by George Wiggans, 1988, USDA) Hash map, then hash each read (or its reverse complement) Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (7) Paul VanRaden
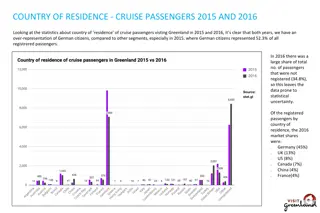
Semi-simulated data l Simulated from UMD3.1 reference map l Variant file from run5 of 1,000 bull genomes w 38,062,190 SNPs, 532,179 insertions, and 1,127,620 deletions l Paired ends, length 150, fragment size 1,000 l Advantage of semi-simulated: true locations and true variants are known Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (8) Paul VanRaden
Compare BWA and findmap Computation required BWA / GATK findmap / findvar 11 2 46 46 CPU minutes / 1X, 1 thread CPU minutes / 1X, 10 threads Memory (Gbytes), 1 thread Memory (Gbytes), 10 threads Variant calling CPU / 1X, 1 thread Accuracy Correctly placed segments overall Both ends of pair correctly placed Both ends wrong 629 63 4.6 46 201 1 90.5% 87.2% 6.2% 92.9% 87.6% 1.8% Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (9) Paul VanRaden
Parallel processing speedup 25 20 15 Optimal IBM3850 HP580 10 5 0 1 2 3 4 5 6 7 8 9 1011121314151617181920 Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (10) Paul VanRaden
Program series example resources Task (per animal) Program Threads Minutes Simulate 10X data Align 10X data and call previous variants map2seq.f90 10 5 findmap.f90 10 20 Sum new variants findvar.f90 1 8 Imputation (39 million) findhap4.f90 10 1 Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (11) Paul VanRaden
Accuracy of variant calling / discovery Known variants SNP (%) 99.8 99.8 86.6 Insertion (%) 98.6 99.8 82.2 Deletion (%) 99.8 99.9 83.7 Correct reference allele Correct alternate allele Call rate (paired ends ok) New variants (Homo / het) Correctly detected (10X) Falsely detected (10X) 91 / 63 10 54 / 37 17 41 / 27 8 Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (12) Paul VanRaden
Other alignment tests l Perfectly random genome, non-repetitive w Over 99.9% correctly aligned l RepeatMasker and BWA w Took 4.4 instead of 14.1 hours / 1X w Only 45% correctly aligned instead of 91% l Human genome gave results similar to cattle Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (13) Paul VanRaden
File format sizes (Mbytes) Unzipped / zipped file sizes BWA, GATK Findmap, Findvar Input data: Sequence reads / 1X (fastq) Output data: Binary alignment file / 1X (.bam) Called genotypes / animal (.vcf) 6000 / 1800 6000 / 1800 3200 / 3200 1200 / 360 1000 / 38 79 / 13 Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (14) Paul VanRaden
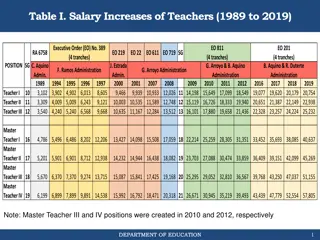
USA use of 1,000 bull genomes l Sequence genotypes from 440 Holsteins l Imputed for 27,000 reference bulls l 700,000 candidate loci + 300,000 HD SNPs l Largest 17K added to 60K routinely used l Average gain of 2.7% reliability across traits l Largest 5K added to next Zoetis chip Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (15) Paul VanRaden
Largest genomic databases 23andMe Ancestry .com CDCB / USDA Genotypes Species Countries Genotyping cost Delivery (weeks) DNA generations EBV reliability Reference: Web sites: >1 million 1.2 million 1.2 million Human Human 55 $199 $99 6-8 6-8 Few Few Low Low http://genomemag.com/davies-23andme/#.VdY722zosY1 https://www.23andme.com/ http://dna.ancestry.com/ https://www.cdcb.us/ http://aipl.arsusda.gov/Main/site_main.htm Cattle 49 $37-135 1-2 Many High Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (16) Paul VanRaden
Conclusions l Program findmap.f90 uses known variants w Alignment is 50X faster than BWA with 1 processor, 30X faster with 10 processors w 2% more segments are mapped correctly w Output files are simpler and 3-10X smaller l Simulation, alignment, variant calling, and imputation programs available from: http://aipl.arsusda.gov/software Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (17) Paul VanRaden
Acknowledgments This research was part of USDA-ARS project 1265-31000-096-00, Improving Genetic Predictions in Dairy Animals Using Phenotypic and Genomic Information. Jeff O Connell provided much advice on alignment and variant calling methods. The reference map was from U. Maryland The variant list was from Daetwyler et al. Plant & Animal Genome, San Diego, California, January 9 -11, 2016 (18) Paul VanRaden













")