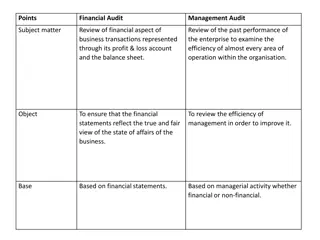
Importance of Internal Audit Functions in Organizations

Internal Audit functions are crucial for providing assurance and management control, especially for medium to large companies, banks, and financial institutions. They help in identifying operational redundancies, serving as an early warning system, and increasing organizational accountability. The International Professional Practices Framework (IPPF) outlines standards for Internal Auditing, emphasizing independence, objectivity, and adding value to organizational operations.

Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
You are allowed to download the files provided on this website for personal or commercial use, subject to the condition that they are used lawfully. All files are the property of their respective owners.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.
E N D
Presentation Transcript
GLP and GCP Chapters 6 & 7 Fundamentals of US Regulatory Affairs 7thEdition
Good Laboratory Practice (GLP) Regulations Chapter 6
Objectives Review the history and purpose of GLP regulations Learn the key GLP regulation components Examine the implications of noncompliance
History of GLPs (Why?) Two Famous Examples of FDA Inspections of Toxicology Studies G.D. Searle and Company Questionable test data Sloppy work Untrained personnel Poor data collection Poor analysis Poor review and reporting practices Industrial Bio-Test Laboratories (IBT) Scientific misconduct Fabrication of Data Replacement of dead animals with healthy ones Changes in the Interpretation of Histopathology Slides Changes in Report Conclusions to make them look more favorable Still goes on today!!!
Introduction The FDA requires manufactures of human drugs, biopharmaceuticals and biological products, animal drugs, medical devices, electronic and food additives to demonstrate the safety of their products prior to use in humans or in the case of animals , prior to use in the indicated species (for the rest of this review, - the manufacturer of will be referred to as drugs ) Compliance with 21 CFR Part 58 the Good Laboratory Practice (GLP) regulation is intended to assure the quality and integrity of the safety data used to support the application s for research or marketing permits for FDA regulated products. GLP Compliance allows for accurate reconstruction of the conduct of a nonclinical study based on quality record keeping and reporting.
GLP Regulations Vary with Agencies and Nations FDA - FD&C Act 1938 and 21CFR58 FDA is currently revising and updating US EPA TSCA (40CFR792) and FIFRA (40CFR160) Applies to chemical substances, mixtures, pesticides OECD (North America, Europe and Asia) EU Requirements Similar to FDA GLPs but includes EPA compounds MHLW (Japanese FDA) Similar to FDA GLPs Mutual Recognition Agreement US FDA, EU and Japanese FDA generally accept each others GLPs (report should state that it meets all 3 agency GLPs) However they do not accept Chinese GLPs FDA, EPA and MHLW GLPs are regulations and required by law, OECD GLPs are voluntary Requirements for all 3 should be met in the Toxicology and Safety Pharmacology reports
GLPs prevent Fraud GLPs were designed to ensure data quality and to reduce fraud by requiring specific documentation be kept regarding several key areas including: Laboratory staff Facilities and Operations Equipment Test and Control Articles Protocol Conduct of study (what happened) Quality Assurance Achieving of data, records and specimens Specific Retention Time required for all for the above for marketed drugs (different for discontinued drugs).
Definition of a Study (1/4) A nonclinical laboratory study means in vivo or in vitro experiments in which test articles are studied prospectively in test systems under laboratory conditions to determine their safety (not in the clinical trials or field trials in animals) .
Types of GLP Studies (2/4) Contract Research Organization or In-house Studies: Acute, Subchronic, Chronic Studies Carcinogenicity and Genotoxicity Studies Development and Reproductive Toxicology Studies Dermal/Eye/Venous/Muscle Irritation Studies Immunogenicity/Antigenicity Studies Bioanalytical Studies of Samples from Dose Groups of Study Animals (Pharmacokinetics/Toxicokinetics) Validation of analytical methods for GLP studies (TK and formulation analysis) Safety Pharmacology Studies (Cardiovascular, Respiratory and CNS)
External Types of Studies (3/4) Laboratories that analyze samples that are generated by GLP studies, they do not conduct the study (can be separate from the laboratory that conducts the in life study): Clinical Chemistry Hematology Urinalysis Histology and Pathology Toxicokinetics Validation of analytical methods for GLP studies (TK and formulation analysis) Immunogenicity Assays
Types of Studies Not Covered by GLP (4/4) Basic Research (Primary and Secondary Pharmacology) Proof of Concept Studies Exploratory Pharmacokinetic Studies (DMPK) Dose-Range Finding Studies (Toxicology) Primary Pharmacodynamic Studies Analytical Quality Control Testing for Clinical and Commercial Studies Stability Testing of Clinical and Commercial Products Studies using Human Studies Field Trials in Animals
FDA GLP Subparts and Sections of 21CFR58 (1/10) (Subpart A General Provisions) Subpart A 58.1 Scope 58.3 Definitions 58.10 Applicability to Studies performed under Grants and Contracts 58.10 Inspection of a testing facility Subpart A 58.3: Definitions Act Test Article Control Article Nonclinical Laboratory Study Applications for Research or Marketing Permit IND / NDA / BLA Notice of Claimed Investigational Exemptions for a New Animal Drug / New Animal Drug Application Application for Premarket Approval of a Medical Device / Product Development Protocol for a Medical Device Sponsor Testing Facilities Person Test System Specimen Raw Data Quality Assurance Unit Study Director Batch Study Initiation Date Study Completion Date
FDA GLP Subparts and Sections of 21CFR58 (2/10) (Subpart A General Provisions) Subpart A: 58.10 Application to Studies Performed under Grants and Contracts Applies to CROs to perform all or part of a nonclinical laboratory study submitted to the FDA as part of an application Sponsor is responsible to tell the CRO the studies are GLP Protocol includes a statement about the regulatory standards under which the study will be conducted (FDA, OECD, EPA, MHLW) Implied that the site should be inspected (audited) by the sponsor if never inspected by the FDA
FDA GLP Subparts and Sections of 21CFR58 (3/10) (Subpart B Organization and Personal) 58.29 - Personnel (Job Descriptions / Training Records) 58.31 - Testing Facility Management Responsible for approving a test facilities Standard Operating Procedures (SOPs) Assigning a Study Director prior to initiation of study and replacing as necessary Responsible for Test and Control Articles Make sure the study complies with GLP 58.33 - Study Director Overall responsibility for the study s design, conduct, reporting and compliance with GLPs and SOPs Document and assess all circumstances and if their impact on the study and corrective actions Not the principle investigator/ contributing scientist if the study is conducted over different sites 58.35 - Quality Assurance Unit Responsible for ensuring that studies are conducted in accordance with GLPs Is independent of the study Maintain a master schedule of GLP studies and inspect the CRO accoding to the schedule as defined by SOPs to insure GLP compliance Responsibility Audit everything - see Subpart A 58.3
FDA GLP Subparts and Sections of 21CFR58 (4/10) (Subpart C - Facilities) Think SOPs! 58.41 General 58.43 Animal Care Facilities 58.45 Animal Supply Facilities 58.47 Facilities for handling test and control articles 58.49 Laboratory Operation Areas 58.51 - Specimen and Data Storage Facilities
FDA GLP Subparts and Sections of 21CFR58 (5/10) (Subpart D - Equipment) 58.81 Equipment Design Computer systems are not covered under GLPs but are included in an FDA inspection and must insure the quality and integrity of the study 21CFR11 Electronic Records and Electronic Signatures established the criteria the FDA uses for electronic records, electronic signatures and hand written signatures executed to electronic records to be trustworthy 58.63 Maintenance and Calibration of Equipment SOPs establish the maintenance and the frequency of calibration of all equipment (scales to air handelers)
FDA GLP Subparts and Sections of 21CFR58 (6/10) (Subpart E Test Facility Operation) 58.81 Standard Operating Procedures (SOPs) Intended to ensure that laboratory procedures ad processes are consistently performed by all individuals in the facility Must be available to all staff anywhere in the facility Mandatory review of all SOPs as needed by Facility Management The protocol describes what needs to be done, SOPs describe how All deviations from SOPs need to be documented in the records and report by the study director and the impact on the study assessed If specific instructions in the protocol are differnet than the SOP, then this is a deviation of the SOP and needs to be documented 58.83 Reagents and Solutions 58.90 - Animal Care
FDA GLP Subparts and Sections of 21CFR58 (7/10) (Subpart F Test and Control Articles) 58.105 Test and Control Article Description Test article - is any food additive, color additive, drug, biopharmaceutical, biologic product, medical device or electronic prodict intended for human use Control article any article other than the test article intended to be used as a control in the study GLPs require the characterization, stability testing sample retention and inventory of test and control articles 58. 107 Test and Control Article Handling Requires the CRO to document the proper storage, handling, distribution, identification, receipt and distribution of the article, showing the chain of custody (think SOPs) 58.113 Mixtures of Articles with Carriers Analytical methods to demonstrate the article is consistently mixed with the carrier (usually sampling from the top, middle and bottom of the mixture is sufficient.
FDA GLP Subparts and Sections of 21CFR58 (8/10) (Subpart G Protocol for and Conduct of a Nonclinical Study) 58.120 Protocol A formal plan of the study 58.130 Conduct of a Nonclinical Laboratory Study The execution, changes to and deviations during the study need to be documented and the impact assessed and recorded. All records in ink and any changes require initials and date of the change. General a single line through the data.
FDA GLP Subparts and Sections of 21CFR58 (9/10) (Subpart J Records and Reports) 58.185 Reporting of Nonclinical Laboratory Results Standard list of what is in the final report FDA inspections of reports follow the Compliance Program 7348.808. GLP statement not required but implicitly required, covered under 21CFR312.23(a)(8)(b)(iii) Investigational New Application and 314.50 (d)(2)(v) Application for FRA approval to market a New Drug 58.190 Storage and Retrieval of Records and Data Establishment of an Archive and Archivist for the storage of all raw data, documentation, protocols, specimens, and interim / final reports and kept in a secure storage area in accordance with 58.195 58.195 Retention of Records
FDA GLP Subparts and Sections of 21CFR58 (10/10) (Subpart K Disqualification of Testing Facilities) 58.200 - Purpose 58.202 Ground for disqualification 58.204 Notice of and opportunity for hearing on proposed disqualification 58.206 Final Order of Disqualification 58.210 Actions upon Disqualification 58.213 Public disclosure of information regarding disqualification 58.215 Alternatives or additional action to disqualification 58.217 Suspension or termination of a testong facility by a sponsor 58.219 Reinstatement of a disqualified testing facility
Proposed Changes to GLP In December of 2010 the FDA issued a public request for comments on proposed revisions to the GLPs
FDA Inspection of GLP Laboratories and the Consequence of Noncompliance All laboratories in the USA can be inspected by the FDA. The FDA needs to request foreign laboratories to conduct GLP inspections. Bioresearch Monitoring Program (BIMO) to address both domestic and international inspections of nonclinical laboratories operating under US GLPs (Compliance Program Guidance 7348.808) 483s (Inspection Observations) Warning Letters Consent Agreements Disqualifications
Good Clinical Practice (GCP) Chapter 7
Objectives Overview of GCP for drugs and devices Overview of clinical trial monitoring responsibilities Differences between HIPAA: FDA s Common Rile and HHS Privacy Rule Overview of Drug and Device Regulations Distinguish Clinical Trial Requirements at the State and Federal Level Provide Regulator Compliance Guidance
Laws, Regulations and Guidelines Lots of them - CFRs, Guidances, ICH (Chapter references 67 of them) GCP are international ethical and scientific quality standards used for designing, conducting, monitoring, auditing, analyzing, recording and reporting clinical trials involving participation of human subjects. Compliance insures the public that the study subject s rights, safety and well-being are protected and consistent with the ethical principles originating in the Declaration of Helsinki Primary Objectives: Protect human subjects during clinical trials Protect patients who might receive an approved product in the future.
Clinical Trials Conduct and GCPs GCPs are covered in the US by 21CFR, Guidances, laws, regulations at the state and federal levels and cover Clinical Trial Responsibility Institutional Review Boards Investigator Obligations US Agencies with GCP regulations include: DHHS Office of Research Integrity FDA 21CFR 11, 50, 54, 56, 58, 312 and 314 NIH Office for Human Research Protections Office of Human Subjects Research and Department of Clinical Bioethics National Human Genome Research Institute International - ICH E6: Guidelines for Good Clinical Practice EU - EMA - Clinical Directive (2001/20/EC)
GCPs primarily apply to the IND The submission of an IND is required prior to conducting a human clinical trail in the US for an unapproved new drug or a new use for a marketed drug. Key personnel are defined by 21 CFR 50 and 312. Sponsor: a person who initiates a clinical investigation but does not conduct the investigation CRO: a person or corporation or agency acting as an independent contractor for the sponsor and who assumes one of more of the sponsor s obligations Sponsor-Investigator: an individual who both initiates and actually conducts, alone or with others, a clinical investigation
Clinical Trials Responsibilities Sponsors Obligations Primary Responsibility -Focus on the clinical study s ethical, scientific and regulatory obligations to protect each study subject s rights, safety and welfare Ensure the proposed clinical trail is adequately supported by nonclinical and clinical information on the investigational product Ensure all clinical trial study personnel are adequately qualified and trained Ensure the protocol is approved by the IRB or Independent Ethics Committee (IEC) Ensure all decisions made on behalf of study subjects are made by a qualified physician Ensure all clinical tasks are performed by qualified personnel Ensure all clinical trials information is recorded Ensure all informed consents are obtained and freely given Ensure the confidentiality records are protected Ensure the investigational product is prepared under cGMPs Ensure quality systems are in place and have been implemented Report unanticipated adverse events
Institutional Review Board (IRB) Obligations The IRB [IEC, Ethics Committee, Independent Research Committee (IRC) or Research Ethics Board (ERB)] protects the rights and welfare of human subjects in clinical trials (21CFR56). Each trial (domestic or foreign) must get IRB approval prior to commencement of the trial Ensure risks to subjects are minimized and reasonable in relation to anticipated benefits Ensure patient selection is as equitable as possible relative to demographics and the clinical trial s purpose Review of protocol, informed consent form (ICF), and Investigator s Brochure (IB) Ensure a pediatric clinical trial is conducted according to regulations Provide initial and continuing review of the ongoing clinical trial at different intervals Request more information if required Approve, disapprove and/or terminate a trial
Investigator Obligations Primary Responsibility is to ensure the rights, safety and welfare of the study subjects Follow the protocol and its specific procedures Ensure the trial s proper conduct at the sites under their authority are in compliance with the signed agreement. The investigational plan, the protocol and all applicable FDA and other government regulations Prepare timely and accurate reports to the IRB, sponsor, medical monitor and FDA if acceptable Maintain accurate records Control the investigational new drug product Ensure that patients sign the ICF before the study procedures begin Maintain appropriate and scheduled communication with the study sponsor, medical monitor and IRB
HIPPA (Health Insurance Portability and Accountability Act Privacy Rule) For the use and disclosure of protected health information for research purposes Established in 1996 and states the minimum federal requirements for protecting the privacy of individually identifiable health information. Define how Protected Health Information (PHI) can be used in clinical trials Health status, health care, payment history, name, address, phone/fax number date of birth email address, SS#
Common Rule Federal Policy for the Protection of Human Subjects Codified under 45CFR46, subpart A and 21CFR 50 and 56. Consent to participate in the research study as a whole not simply for the research use or disclosure of protected health information
GCPs for Device Trials (cover under medical devices) Are different than drugs ICH E6 states that an FDA form 1572 must be submitted for a drug trial It does not have to be submitted in a medical device trail, however, the sponsor should provide the elements listed under 21CFR812.43*c) to the investigator and the principle investigator (PI) should sign this Investigator Agreement prior to commencing a device clinical trial. ISO - Internal Standard 13155:2011, Clinical investigation of Medical Devices for Human Subjects Good Clinical Practice applies
Clinical Trial Regulations Designed to ensure that clinical trial subjects are protected from harm, that the subjects rights are protected and that the trial s scientific integrity is assured. They require: A strong, effective and efficient regulatory framework Willingness of investigators to participate and comply Adequate site/research infrastructure Willingness of patients to participate and comply Differ between drugs and Devices Differ at the Federal and State Level
Federal Clinical Trial Requirements - Differences Regulation Drugs Devices IND/IDA 21CFR 312 21CFR 812 IND/IDE Application Process 21CFR 312.23 21CFR 812.20 and 812.25, 27 Form FDA 1571 21CFR 312.23(a)(1) Form FDA 1572 21CFR 312.53(c) 21CFR 812 .43(c) not required for devices Foreign Clinical Trials not conducted under an IND 21CFR 312.120 Legal Representation 21CFR 321.52 Labeling 21CFR 312.6 21CFR 812.5 Responsibilities of Sponsors and Investigators 21CFR 312 subpart D 21CFR 812 subpart C and E Records and Reports 21CFR 312, 57, 58, 62, 64, 68 21CFR 812.140 and 150 Safety Reporting 21CFR 312.32 21CFR 803 Bioavability and Bioequivalence Requirements 21CFR 320 Quality System Regulations 21CFR 820
Clinical Trial Regulations Applicable to IND and IDA Regulation Source Electronic Records; Electronic Records 21CFR11 Protection of Human Subjects (Informed Consent) 21CFR50 Financial Disclosure by Clinical Investigators 21CFR54 IRBs 21CFR56 Form FDA 3674 (Certification) Food and Drug Administration Amendment Act of 2007 HIPPA Privacy Act HIPPA Pediatric Clinical Trials 21CFR50 Subpart D Additional safeguards for Children in Clinical Investigations
State Clinical Trial Requirement Compliance with individual state clinical trial regulations is one of the most difficult compliance issues for a sponsor as state requirements may differ form federal regulations. Same holds true for international clinical programs Need legal advice for different states or countries
State Clinical Trials Insurance Requirements About half of the state legislatures have passed clinical trial legislation and /or instituted special agreements requiring health plans to pay the cost of routine medical care for study subjects Table 7-4 lists individual states
Clinical Trial Regulatory Compliance Most noncompliance in clinical trial research is due to clinical trial or protocol misconduct The investigator, the sponsor, the institution and the IRB are responsible for subject safety Enforcement: DHHS(45CFR46) - noncompliance reported to IRB, Institutional officials and agency FDA (21CFR56.120-124) IRB must report to the agency Has enforcement authority [Office of Regulatory Affairs (Bioresearch Monitoring (BIMO)) that does onsite inspections and audits] NIH
Financial Disclosure Anyone submitting a marketing application to the FDA must also submit lists of participating clinical investigators who conducted clinical trials and certify and/or disclose certain financial arrangements
Pharmacovigilance in Clinical Trials Sponsors are required to review all information pertaining to the safety of an IND from any domestic or foreign sources. An IRB is also required to continue to review the study at set intervals to assure the protection of human subjects rights and welfare.
Data Monitoring Committee (DMC) A group of individuals with relevant expertise who regularly review accumulating data from one or more ongoing clinical trials. The DMC advises the sponsor regarding the ongoing safety of enrolled and prospective study subjects and the continuing validity and scientific merit if the trial.
Clinical Trial Noncompliance Common Issues Clinical Trail Monitoring Noncompliance Issues Falsified Data Plagiarism and falsified study results False or misleading protocols Failure to report Unanticipated AE Implementing changes to the protocol or ICF without first submitting it to the IRB Commencing significant changes to a protocol without first submitting for FDA approval Enrolling a study subject who does not meet the inclusion criteria Failure to withdraw study subjects who no longer meet inclusion criteria or who meet one or more exclusion criteria Failure to make timely reports to the IRB Research conducted without IRB review
Investigator Noncompliance Issues Unreported changes to the protocol Misuse or nonuse of the Informed Consent Form (ICF) Failure to submit protocols to the IRB in a timely fashion
IRB Noncompliance Issues Inadequate research protocol review Failure to ensure that the ICF and process provide sufficient information to allow prospective subjects to make an informed decision as to whether to participate in the research Failure to ensure that the research design includes adequate data monitoring and any additional safeguards necessary to protect the welfare of particularly vulnerable patients Failure to conduct continuing review of research at intervals appropriate to the degree of risk Failure to maintain adequate records of IRB business Insufficient information to make determinations required for approval of research
IRB Noncompliance Issues (cont) Inadequate review at convened meetings Inadequate continuing review Contingent approval of research with substantive changes and no additional review by the convened IRB Failure to conduct continuing review at least once annually Meeting convened without quorum due to the absence if a nonscientist Meeting convened without quorum due to a lack of a majority Members with conflicting interest participated in IRB review of research
Institutional Noncompliance Issues Failure to ensure that the IRB has the required number of members and functions in accordance with regulations Failure to ensure that the IRB receives appropriate institutional support and staffing Failure to ensue that investigators meet their obligations to the IRB
Regulatory Compliance Sanctions At the DHS Level DHHS regulations di not specify administrative actions for noncompliance other than the following Failure to comply with regulations can result in termination or suspension of support for agency projects. Demonstrated inability to carry out IRB responsibilities in accordance with DHHS regulations can be cause for suspension or withdrawal of approval of an institution's assurance Systemic failure to abide by the terms and conditions or an institution's assurance will result in withdrawal of approval of the assurance.

")

")



")
")
")
")
")
")
")
")
")
")
")
")
")
")


")





 Obligations")













")



")