PRIMARY BILIARY CIRRHOSIS

Primary biliary cirrhosis is an autoimmune liver disease characterized by the progressive destruction of small bile ducts. It can lead to scarring, fibrosis, and cirrhosis. Common symptoms include fatigue, itching, jaundice, and more. Diagnosis involves blood tests, autoimmune screening, radiology, and liver biopsy.

Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
You are allowed to download the files provided on this website for personal or commercial use, subject to the condition that they are used lawfully. All files are the property of their respective owners.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.
E N D
Presentation Transcript
PRIMARY BILIARY CIRRHOSIS PRESENTED BY : Hawa-A-Elbarghthi Marwa-A- Elfazani
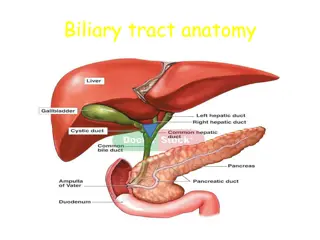
Primary biliary cirrhosis It is an autoimmune disease of the liver by: the slow progressive destruction of the small bile ducts (bile canaliculi) within the liver. when these ducts are damaged, bile builds up in the liver (cholestasis) and over time damages the tissue. this can lead to scarring, fibrosis and cirrhosis.
Epidemiology Female : male ratio of 9:1 Most common during middle age Commonest in northern Europeans, least common in Africans Prevalence: 19-150 cases/million Incidence: 4-15 cases/million/yr First-degree relatives may have as much as a 500 times increase in prevalence
Clinical features Fatigue (most common) Itching (Pruritus) Dry skin and eyes Abnormal bleeding or bruising Jaundice Xanthelasma and xanthomata Hepatosplenomegaly Features of chronic liver disease Palmar erythema; spider naevi; cachexia; ascites Pale stool or dark urine
PBC Differential diagnosis Biliary stones or strictures Pancreaticobiliary malignancies Autoimmune hepatitis Alcoholic hepatitis Viral hepatitis Sarcoidosis Autoimmune cholangiopathy Medications Granulomatous hepatitis
diagnosis of primary biliary cirrhosis Blood tests: Liver function tests elevated alkaline phosphatase ALP and gamma-glutamyl transferase GGT are found in early disease. Elevations in bilirubin occur in advanced disease. Clotting Elevated prothrombin time Full blood count Trombocytopenia if cirrhosis present Serum lipids Cholesterol, LDL and HDL all significantly raised Full liver screen of blood tests to rule out other causes of liver disease (see chronic liver disease section)
Autoimmune screen: Serum antimitochondrial M2 antibodies (95% sensitive, 98% specific) Elevated serum immunoglobulins, especially IgM Radiology Ultrasound liver to look for focal liver lesions, portal/hepatic vein thrombosis, extrinsic causes of biliary duct compression. Magnetic resonance cholangiopancreatography (MRCP) gives a more detailed view of the biliary tree and does not have the associated morbidity of Endoscopic retrograde cholangio pancreatography (ERCP). CT abdomen : This is more likely to be performed if an extrahepatic cause of cholestasis is suspected. Liver biopsy A liver biopsy is necessary to determine the stage of disease.
stages of the disease Stage 1 - Portal Stage: Normal sized triads; portal inflammation, subtle bile duct damage. Granulomas are often detected in this stage. Stage 2 - Periportal Stage: Enlarged triads; periportal fibrosis and/or inflammation. Typically characterized by the finding of a proliferation of small bile ducts. Stage 3 - Septal Stage: Active and/or passive fibrous septa. Stage 4 - Biliary Cirrhosis: Nodules present; garland or jigsaw pattern.
Complications and associations of PBC Complications and associations of primary biliary cirrhosis Osteoporosis Malabsorption of fats and fat-soluble vitamins can lead to osteomalacia and coagulopathies Liver cirrhosis and its complications Renal tubular acidosis Associated conditions Hypothyroidism (seen in up to 20%) of patients Rheumatoid arthritis; Systemic sclerosis; Sjogren s syndrome; Sicca syndrome
Chronic management of primary biliary cirrhosis medical treteamt If PBC is diagnosed at an early histologic stage and treatment with UDCA is begun, recent studies have suggested that the long-term survival approaches that of a healthy control population.* Ursodeoxycholic acid (UDCA) 13-15mg/kg/day in usually two divided dosesCan be used in any patient with PBC and abnormal liver biochemistry. Can significantly improve liver biochemistry and reduce the need for liver transplantation and overall mortality. has no effect on pruritus, fatigue or associated bone disease
Steroids and other immunosuppressive agents Prednisolone has been shown to significantly improve liver biochemistry, however, it makes bone disease much worse and thus is not recommended long-term Steroid-sparing immunosuppressant drugs have not been shown to be effective in PBC
Anti-pruritics :Cholestyramine: 4g per dose up to 16g/day given 2-4 hours apart from UDCA. Rifamipicin at a dose of 150mg once or twice daily Opiate agonists such as Naltrexone at a starting dose of 50mg daily Surgcial Orthotopic liver transplantationIs indicated in patients with end-stage disease Patients should be referred when the bilirubin level is > 100 micromls/litre or earlier if debilitating symptoms. Up to 20-25% of patients will have disease recurrence at 10 years post-transplant
Prognosis of primary biliary cirrhosis Some studies suggest that asymptomatic patients have a 50-70% 10 year survival whereas median survival from onset of symptoms is 5-8 years. The serum bilirubin level is a marker of prognosis