Understanding Urea Cycle Disorders and Hyperammonaemia: Overview, Diagnosis, and Treatment
Dive into the world of urea cycle disorders, a genetic condition resulting from enzyme deficiencies, causing hyperammonaemia. Learn about the urea cycle, regulation, clinical features, lab investigations, and treatment options for this rare disorder.

Download Presentation

Please find below an Image/Link to download the presentation.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author. If you encounter any issues during the download, it is possible that the publisher has removed the file from their server.
You are allowed to download the files provided on this website for personal or commercial use, subject to the condition that they are used lawfully. All files are the property of their respective owners.
The content on the website is provided AS IS for your information and personal use only. It may not be sold, licensed, or shared on other websites without obtaining consent from the author.
E N D
Presentation Transcript
Introduction Genetic disorder caused by mutation which results in deficiency of one of the five enzyme C/F: 1 in 30000 live births Hyperammonaemia Encephalopathy Respiratory alkalosis
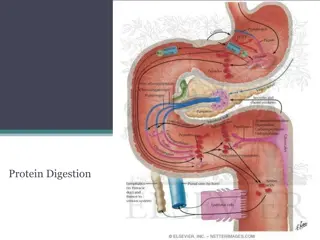
Urea Cycle Krebs Henseleit cycle / Ornithine cycle Urea is formed from Ammonia, CO2 and Aspartate Site: Liver Subcellular Site: Mitochondria / Cytosol
Ammonia is trapped as Glutamine Brain & Kidney small amount of Ammonia is generated and trapped Ammonia is Toxic Normal concentration- 10-20microgm/dL
Regulation of Urea Cycle Coarse Regulation: 1. Starvation 2. Protein rich diet Fine Regulation: 1. Allosteric regulation- NAG activates CPS I Compartmentalization: 1. 1sttwo enzymes are in Mitochondria 2. Rest in cytoplasm
Hyperammonaemia Type 1 Rare and characterized by severe hyperammoneamia Defect- CPS 1 Autosomal recessive Gene 2q35 Incidence : 1 in 100,000 C/F: MR
NH3 + CO2 Carbamyl phosphate CPS NAG
Clinical Features 1. Refusal to eat 2. Vomiting 3. Lethargy 4. Convulsion 5. Coma
Lab investigations Hyperammonaemia without increase in specific AA Increased glutamine and alanine secondary to Hyperammonemia Urine orotic acid low / nil
Treatment Oral administration of Carbamylglutamate
Hyperammonaemia Type 2 X linked trait Hemizygous males than heterozygous female Defect- OTC
Clinical Manifestation 1. Ataxia 2. Mental confusion 3. Vomiting 4. Agitation 5. Occurs after ingestion of High protein Diet
Lab investigations 1. Hyperammonemia without increase in any specific AA 2. Increase in glutamine and alanine secondary to hyperammonemia 3. Increased in Urinary orotic Acid 4. Mimic lysinuric protein intolearnce
Identification of Mutant gene Pre natal- Liver biopsy DNA analysis from Chronic Villous Samples
Treatment Liver transplantation
Citrullinemia Autosomal Recessive Defect- Arginosuccinate synthase 1. Type I 2. Type II
Citrulline + aspartic acid ASA ASA synthetase
Type I citrullinimia 1. Severe / Neonatal form- first few days of Life 2. Subacute / mild form FTT, frequent vomiting, Developmental Delay, dry and brittle Hair Gradually after 1yr of Life AR 9q34
LAB: Plasma citrulline increased Urine orotic acid increased
Prenatal CVS DNA analysis Enzyme activity TREATMENT: 1. Arginine 2. Prognosis is poor 3. Protein restricted diet with sodium benzoate and arginine
Type II citrulinimia Defect: Citrin SLC25A13 7q21.3 Transport Aspartate from mitochrondria to cytoplasm 2 types 1. Type II neonatal form 2. Type II adult form
Type II neonatal form Known as neonatal hepatic cholestasis 3months of age Include jaundice with mild hyperbilirubinemia Hypoproteinimia Increased PT Increased ALP
Lab Investigation Plasma ammonia and citrulline normal Increased methionine, tyrosine, alanine Increased AFP Liver biopsy: fatty infiltration, cholethiasis with dilated canaliculi, fibrosis Seen in JAPAN
Type II Adult form Disorientation Delirium Delusion Tremor Frank psychosis 20-40yrs Liver transplantation
Arginosuccinate Aciduria Defect- arginosuccinate Lyase AR 7cen q 11.2 1stfew days of Life Mortality high Dryness and brittle Hair ( trichorrechexis nodosa)
Arginosuccinic Acid Arginine ASA lyase
Lab Investigation 1. Increased Liver enzymes 2. Increased Glutamine & alanine 3. Increased Citrulline 4. ASA found in Urine and CSF
Hyperarginemia Mild variety 2-4yrs of age Defect- arginase AR Two enzymes 1. Cytosolic A1( liver and erythrocyte) 2. Renal and brain mitochrondria (6q23)
Arginine ornithine Arginase
Clinical Features Rare Insidious onset Remain asymptomatic Progressive spastic diplegia with scissoring lower extremities Developmental delay MR Seizure
LAB INVESTIGATION Increased arginine Urine orotic Acid increased Diagnosis: arginine in urine Treatment: 1. Low protein diet 2. Adm of synthetic protein made of essential AA 3. Sodium benzoate
HHH syndrome Defect- ornithine transporter AR SLC25A15 Hyperornithinemia, hyperammonemia, homocitrullinemia Transport system from cytosol to mitochondria Accumulation of ornithine in cytosol Defn of ornithine in mitochondria
Clinical features Vomiting Lethergy Refusal to feed Increased DTR Seizures Clonus
Lab Investigation Increased plasma ornithine Increased plasma Homocitrulline
Transient Hyperammonaemia of New Born Very low birth infant mild which last for about 6-8weeks Asymptomatic follow up to 18months Severe transient hyperammonaemia- observed in New born infant with premature and respiratory distress
Acquired Hyperammonaemia Liver cirrhosis Loss of hepatocytes Replaced by fibrous connective tissue Impairs the blood flow
Ammonia Estimation Formed from deamination of AA during protein metabolism Removed from circulation and converted into urea Free ammonia is toxic and present in plasma in low concentration
Physiology NH3 is formed from the catabolism of AA & bacterial metabolism in Lumen of Intestine NH3 consumed by parenchymal cells of Liver to produce Urea Normal physiologic pH it is ammonium ion and excreted by kidney and act as buffer
Monitoring NH3 used in the prognosis Arterial NH3 is the best indicator of the severity of disease Used in 1. Hepatic failure 2. Reye s syndrome 3. Inherited UCD
methods for estimation 1. Chemical methods 2. Enzymatic method